MeRIP-seq for Detecting RNA methylation: An Overview
-
 Loading...
Loading... - kiko Garcia
- 27 Mar 2025
- 36 Views
- 0 Like
- 0 Comment
Explore MeRIP-seq, a key technique for detecting RNA methylation. Learn about its workflow, advantages, limitations, and its applications in gene expression research.
Epitranscriptome pertains to the phenomenon wherein gene expression is modulated by chemical alterations on RNA without altering the RNA sequence. There exist over 100 types of intracellular RNA modifications, with the majority being conspicuous modifications on tRNA and other non-coding RNA, and only a few modifications on mRNA. The most abundant modification in mRNA is N6-methyladenosine (m6A), which partakes in all facets of the mRNA life cycle. Research has demonstrated that the primary function of m6A is to govern the stability of mRNA.
What is MeRIP-seq
Presently, high-throughput sequencing, colorimetry, and liquid chromatography-mass spectrometry (LC-MS) are the technical modalities for detecting m6A. Colorimetry and LC-MS can measure the overall m6A level of mRNA. LC-MS/MS employs tandem mass spectrometry based on liquid mass spectrometry to acquire molecular ion peaks and fragment ion peaks, and concurrently conducts qualitative and quantitative analyses of bases. However, these two methods are incapable of localizing m6A and are typically employed in the initial stages of research. Currently, methylated RNA immunoprecipitation sequencing (MeRIP-seq) is predominantly utilized in high-throughput sequencing and is one of the most prevalently used techniques for investigating m6A modification.
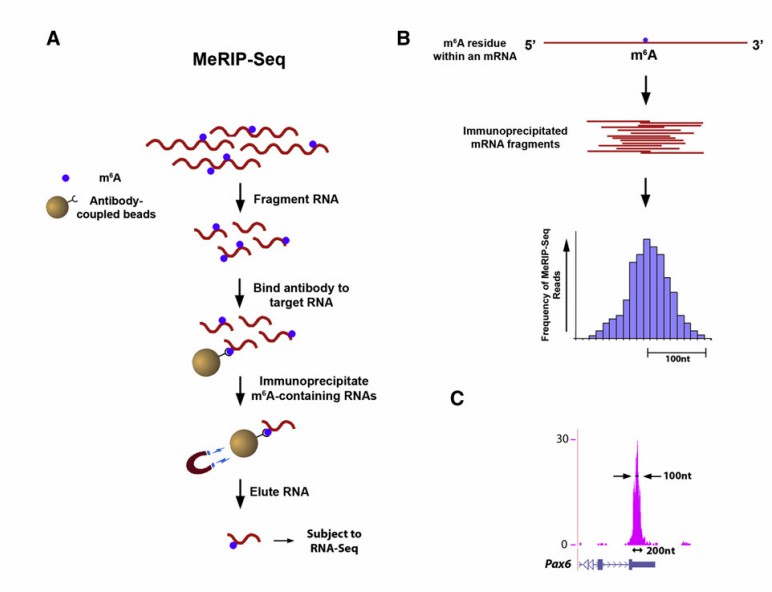
MeRIP-seq amalgamates methylated DNA immunoprecipitation (Medip) technology, RNA immunoprecipitation (RIP) technology, and RNA sequencing (RNA-seq) technology to precisely detect RNA methylation across the entire genome (or whole transcription group). The principle involves the specific recognition of m6A-modified antibodies to immunoprecipitate m6A-modified RNA fragments within cells. Through high-throughput sequencing analysis of the precipitated RNA fragments in conjunction with bioinformatics analysis, the modification of m6A in the entire genome can be systematically examined.
Principle of MeRIP-seq
Immunocoprecipitation: Specific antibodies are employed to identify and bind to RNA fragments possessing specific methylation modifications within cells. For instance, antibodies against m6A modification can recognize and bind to RNA with m6A modification, forming antibody-RNA complexes, which are then bound using magnetic beads and other tools for complex enrichment, thereby enabling the specific separation of methylated RNA from total RNA.
Library construction and sequencing: After the enriched methylated RNA is purified, it is reverse transcribed using a specific kit and method, and a sequencing library is constructed. Subsequently, a large number of short sequence reads are obtained through sequencing on a high-throughput sequencing platform.
Bioinformatics analysis: Quality control and filtering are performed on the original sequencing data, eliminating linker sequences and low-quality bases. The filtered data is then aligned to the reference genome. Owing to the numerous read alignments at methylation modification sites in the sequencing data, "peaks" are formed. By detecting the positions of these peaks, the methylation regions on RNA can be determined, followed by the localization of methylation sites and quantitative analysis of the methylation level, ultimately yielding the map and related information of RNA methylation within the whole transcription group.
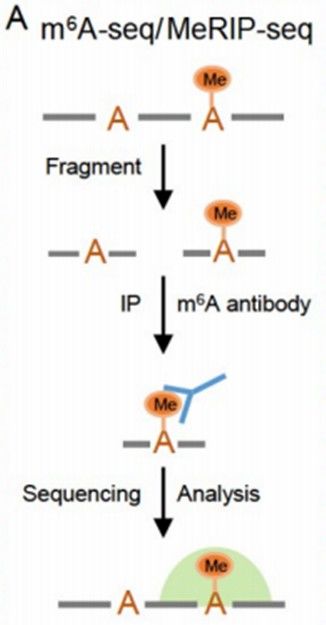
Principle of MeRIP-seq (Yang et al., 2023)
How Does MeRIP-seq Work
MeRIP-seq accurately identifies m6A-modified RNA regions by extracting RNA, selectively enriching m6A-modified RNA, constructing libraries, and performing high-throughput sequencing, providing powerful support for studying RNA modifications and their role in diseases.
Sample preparation: Cell or tissue samples are collected, and total RNA is extracted. Quality inspection is carried out, such as agarose gel electrophoresis to assess the degree and contamination of RNA, which necessitates the presence of distinct 18S or 28S main bands and clear bands. RNA concentration is accurately quantified using Qubit 2.0, with a total amount of not less than 10μg. RNA integrity is precisely detected using Agilent 2100, with an RIN value of not less than 7.5.
Library construction: 10μg of total RNA is taken, and RNA Fragmentation Reagents are added. The reaction is conducted in a Thermomixer at 70℃ for 10 minutes to fragment the RNA into approximately 100nt fragments, which are then precipitated with ethanol. The magnetic beads containing protein A and protein G are washed with IP buffer and incubated with 5μg of m6A antibody at 4℃ for 2 hours. After washing twice with IP buffer, the magnetic beads are resuspended, and the fragmented RNA is added. The mixture is incubated at 4℃ for 4 hours with gentle agitation. Subsequently, the magnetic beads are washed three times with IP buffer at 4℃, incubated with m6A competitive eluent at 4℃ for 1 hour, and the supernatant containing the eluted m6A RNA is collected and purified using phenol chloroform and isoamyl alcohol. IP and Input samples are reverse transcribed and library construction is performed according to the Smarter Stranded Total RNA-Seq Kit V2-Pico Input Mammalian User Manual, and the fragment size is selected using AMPure XP Bead to obtain the final library.
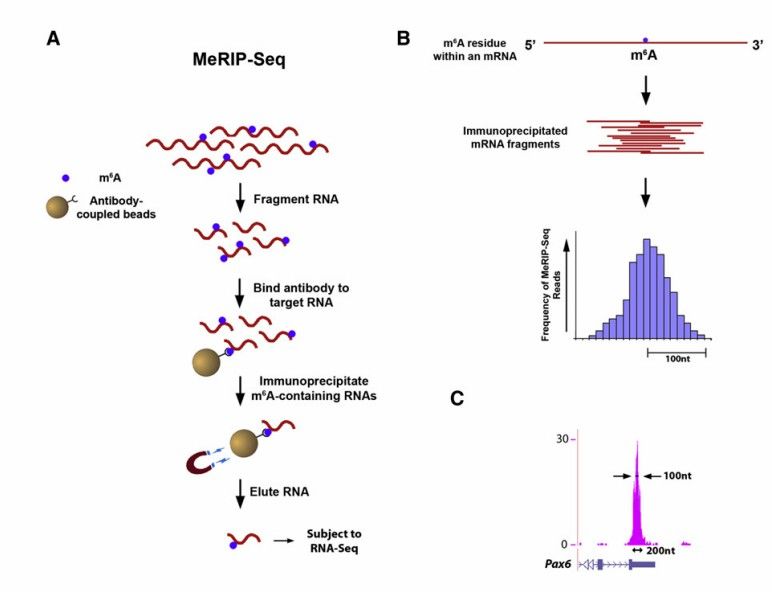
Protocol of Merip-seq (Kate et al., 2012)
Library quality inspection: The library is initially quantified using Qubit 2.0 and diluted to 1 ng/μL. Subsequently, the Insert Size of the library is detected using Agilent 2100. Once it meets the expectations, the effective concentration of the library is accurately quantified using the qPCR method to ensure it is greater than 2nM.
Computer sequencing: After the library is qualified, different libraries are pooled and sequenced on the Illumina Nova platform in accordance with the requirements of effective concentration and target offline data, with a sequencing strategy of PE150.
Detection of m6A modified region: Since the number of reads covering the m6A modified region in the IP library is significantly higher than that in the Input library, a peak is formed, and the position of m6A-modified RNA can be determined by detecting the position of the peak.
How to Analyse MeRIP-seq Data
MeRIP-seq data analysis involves quality control, alignment, detection of methylation sites, identification of differential methylation regions, and functional enrichment analysis, providing insights into the role of RNA methylation in biological processes and diseases.
Data preprocessing: Software such as FastQC is used to assess the quality of the original sequencing data, including sequencing base quality, GC content, and sequence length distribution. If the terminal base quality of the sequencing data is found to be low, Trimmomatic software can be employed to remove low-quality bases and linker sequences to ensure the data quality meets the requirements for subsequent analysis. Commonly used tools like HISAT2 or STAR are used to align the filtered data to the reference genome or transcriptome. For example, when studying human samples, the data is aligned to the reference sequence of the human genome to ascertain the origin location of the sequencing reads.
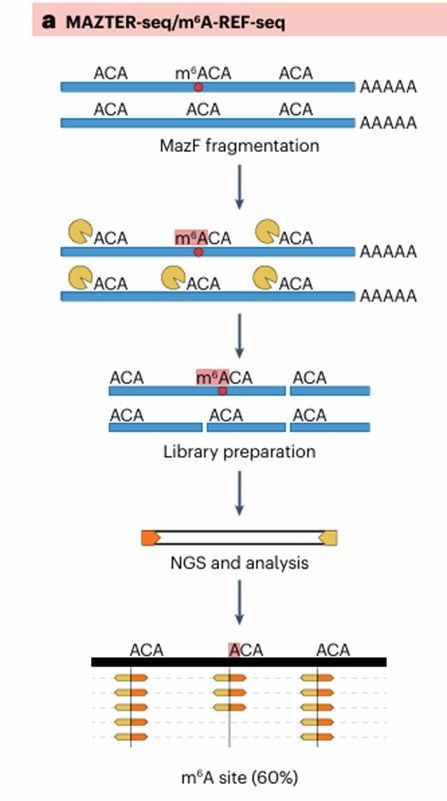
Additional methods for m6A profiling (Michal et al., 2024)
Identification and analysis of methylation sites: Software such as MACS2 is used to detect methylation-rich regions, i.e., peaks, which represent the potential locations of methylation sites. For example, by comparing the sequencing data of immunoprecipitation samples and input samples, the region with a higher enrichment factor is regarded as the potential methylation site region. The methylation level of methylation sites or regions is calculated, generally based on the number of reads covering a specific area in immunoprecipitation samples and input samples to reflect the methylation degree of the site.
Screening differential methylation regions (DMRs): Using software like DiffBind, the methylation levels between different sample groups (such as the disease group and the normal group) are compared, and DMRs are screened out. These DMRs may be associated with the occurrence and development of diseases.
Functional enrichment analysis: The functional enrichment of DMR-related genes is analyzed, often using tools such as DAVID or Metascape to understand the biological processes, molecular functions, and signal pathways involved by these genes. If the related genes are found to be enriched in pathways related to cell proliferation and differentiation, it can be inferred that methylation modification plays a role in these processes.



